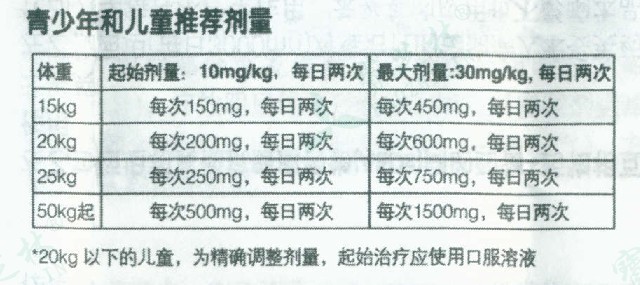
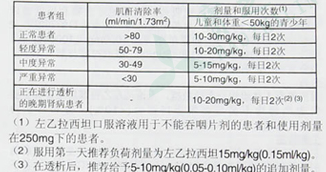
开浦兰 左乙拉西坦片癫痫宝芝林大药房开浦兰 左乙拉西坦片页面提供比利时 UCB Pharma S.A.生产的开浦兰 左乙拉西坦片,开浦兰 左乙拉西坦片说明书涵开浦兰 左乙拉西坦片价格、开浦兰 左乙拉西坦片的功效与作用、开浦兰 左乙拉西坦片的用法用量、开浦兰 左乙拉西坦片的副作用与不良反应、开浦兰 左乙拉西坦片的注意事项与禁忌等信息。





 地址: 广州市越秀区先烈南路21号之五首层112房
地址: 广州市越秀区先烈南路21号之五首层112房
 地址: 广州市越秀区中山二路52号首层(中山医附属第一医院旁)
地址: 广州市越秀区中山二路52号首层(中山医附属第一医院旁)
 地址: 广州市越秀区东川路92号(广东省人民医院旁,东川市场二楼)
地址: 广州市越秀区东川路92号(广东省人民医院旁,东川市场二楼)
 地址: 广州市越秀区龟岗大马路5-9号
地址: 广州市越秀区龟岗大马路5-9号
 地址: 广州市越秀区大德路116号首层(省中医对面)
地址: 广州市越秀区大德路116号首层(省中医对面)
 地址: 广州市白云区白灰场北街1号自编110号铺
地址: 广州市白云区白灰场北街1号自编110号铺
 地址: 广州市海珠区恒信路165-175号之一(广东省第二人民医院,即原解放军第一七七中心医院斜对面)
地址: 广州市海珠区恒信路165-175号之一(广东省第二人民医院,即原解放军第一七七中心医院斜对面)
 地址: 广州市荔湾区芳村大道中316号(恒荔湾畔唯品会楼下)
地址: 广州市荔湾区芳村大道中316号(恒荔湾畔唯品会楼下)
 地址: 广州市荔湾区明心路40号首层
地址: 广州市荔湾区明心路40号首层
 地址: 广州市天河路633号
地址: 广州市天河路633号
 地址: 广州市越秀区靖海路29号一、二楼
地址: 广东省佛山市禅城区普澜公路澜石糖仓大院内第1号房之5号铺
地址: 广州市越秀区淘金路54号(世贸花园之地下西侧建筑)
地址: 广州市海珠区南州北路46号101
地址: 广州市越秀区东川路79号首层及二楼
地址: 广州市越秀区人民北路592-594号一楼及二楼全层
地址: 广州市越秀区中山二路78号首层自编5-7号(省人民医院急诊对面)广东省人民医院
地址: 广州市荔湾区中山八路25号首层103铺(富力商贸大厦首层)
地址: 广州市海珠区建基路98号首层自编1-6
地址: 广州市越秀区靖海路29号一、二楼
地址: 广东省佛山市禅城区普澜公路澜石糖仓大院内第1号房之5号铺
地址: 广州市越秀区淘金路54号(世贸花园之地下西侧建筑)
地址: 广州市海珠区南州北路46号101
地址: 广州市越秀区东川路79号首层及二楼
地址: 广州市越秀区人民北路592-594号一楼及二楼全层
地址: 广州市越秀区中山二路78号首层自编5-7号(省人民医院急诊对面)广东省人民医院
地址: 广州市荔湾区中山八路25号首层103铺(富力商贸大厦首层)
地址: 广州市海珠区建基路98号首层自编1-6
 地址: 广州市海珠区同福中路391号首层
地址: 广州市海珠区同福中路391号首层
 地址: 广州市越秀区人民中路296号[儿童医院院区](面向广州市儿童医院右侧)
地址: 广州市越秀区人民中路296号[儿童医院院区](面向广州市儿童医院右侧)
 地址: 广州市越秀区农林下路14号首层自编之二、之三
地址: 广州市越秀区农林下路14号首层自编之二、之三
 地址: 广州市天河区员村二横路11号大院13号之一自编2号
地址: 广州市天河区员村二横路11号大院13号之一自编2号
 地址: 广州市越秀区中山三路3号首层自编002房
地址: 广州市越秀区中山三路3号首层自编002房
 地址: 广州市白云区三元里街机场路10号之二号1050铺、1051铺、1059铺、1060铺、1061铺、1069铺、1070铺、1071铺、1072铺、1073铺、1074铺、1075铺
地址: 广州市白云区三元里街机场路10号之二号1050铺、1051铺、1059铺、1060铺、1061铺、1069铺、1070铺、1071铺、1072铺、1073铺、1074铺、1075铺
 广州市天河区金穗路34号首层1号铺之三
广州市天河区金穗路34号首层1号铺之三
 广州市番禺区南村镇兴南大道486号110房、111房、112房、113房
广州市番禺区南村镇兴南大道486号110房、111房、112房、113房
 广州市白云区云景路祥云街7号4、5、26、27房
广州市白云区云景路祥云街7号4、5、26、27房
 广州市白云区嘉禾街望岗德兴路105号
广州市白云区嘉禾街望岗德兴路105号
 广州市天河区黄埔大道872号首层108号铺
广州市天河区黄埔大道872号首层108号铺